










Happenings @yenepoya

Valedictory Ceremony of 'Icon Youth 2025- International Conference' at Yenepoya (Deemed to be University)
19 May 2025

The Honorable Delegates from S Vyasa University, Bangalore Dr. H R Nagendra -Chancellor, Dr. N.K. Manjunath - Vice Chancellor, Dr. H R Dayananda Swamy - Director - Finance & Administration, and Dr. Amit Kumar Singh During their Visit to the Yenepoya (Deemed to be University), Mangalore.
16 May 2025

The two-day international conference, ICON YOUTH 2025, themed "Envisioning India's Future: Youth as Change Makers", was inaugurated today at the Yendurance Zone of Yenepoya (Deemed to be University), Mangaluru. Jointly organized by the Ministry of Youth Affairs and Sports, Government of India, and Yenepoya (Deemed to be University)
16 May 2025

Portable Ultrasound Machine was Handed over to Zulekha Yenepoya Institute of Oncology by Mr. Padmanabhan Desikachari, Founder, IndyGen Labs & Chairman of Appnomic Systems.
07 May 2025


Yenepoya (Deemed to be University) hosted an exhibition stall highlighting our Educational and Healthcare services at the Karnataka Beary Festival held at the Karavali Utsava Ground on 19th and 20th April 2025.
22 Apr 2025


Yenepoya Deemed-to-be University NSS Unit organized a Blood Donation Drive and University NSS Level Awards to honor the Best NSS Program Officers & Volunteers for their exemplary service to the community
24 Mar 2025

Yenepoya Medical College Hospital Celebrates International Women's Day & World Kidney Day
18 Mar 2025

Yenepoya (Deemed to be University), Gender Sensitization Cell (GSC) observed 'International Women's day on 08.03.2025 with the theme "ACCELERATE ACTION"
08 Mar 2025

The National Science Day 2025, Organised by the Yenepoya (Deemed to be University)
01 Mar 2025

The first-ever edition of Racefor7 in Mangaluru, organized by the Organization for Rare Diseases India (ORDI) in association with Yenepoya (Deemed to be University) and Arunya Foundation, concluded successfully recently (last Sunday) at Yendurance Sports Zone
26 Feb 2025

2-day workshop on Scientific Writing 10-11 February 2025 and Career Counseling in Clinical Ethics 7 February 2025 by The Centre for Ethics, Yenepoya (Deemed to be University) featuring esteemed Prof. Henry Silverman, University of Maryland, USA
13 Feb 2025

Yenepoya (Deemed to be University), Launched the Calendar 2025, during the Sneha Milana 2024 event
24 Dec 2024



Department of Hospital Administration, Yenepoya (Deemed to be University), in Collaboration with CAHO, Successfully Conducts CPQH Workshop
12 Nov 2024






Report on the Valedictory Programme of the Five-Day National Level Workshop on "Econometric Analysis in Leading Academic Journals: Hands on Practice"
28 Sep 2024

Exploring the Research Constituents: A Guide to Export Data and Conduct Bibliometric Analysis using VOSviewer
28 Sep 2024




Rashtriya Poshan Maah 2024 Launched with Enthusiasm at Yenepoya Naturopathy and Yogic Science College and Hospital
02 Sep 2024

Mangaluru: Installation of Student Council 2024-25 held at The Yenepoya Institute of Arts, Science, Commerce and Management, Kulur Campus
02 Sep 2024

Yenepoya Medical College Hospital Pledge to Prevent Mosquito Borne Diseases on
20 Aug 2024



Breastfeeding Day Celebration by the NURSING SERVICE DEPARTMENT of YENEPOYA MEDICAL COLLEGE HOSPITAL
10 Aug 2024

Report on symposium titled "Aligning Grant Proposals with Sustainable Development Goals (SDG) for Viksit Bharat"
10 Aug 2024

Yenepoya (Deemed to be University) Offers Free Education to Students From Families Affected by the Wayanad Disaster
07 Aug 2024

Valedictory Ceremony of International Conference on Mixed Method Research: Integrating Insights and Advancing Healthcare
03 Aug 2024

International Conference on Mixed Method Research: Integrating Insights and Advance Healthcare
02 Aug 2024

Inaugural Ceremony of International conference on Mixed Method Research: Integrating Insights and Advancing Healthcare
02 Aug 2024




Report on School Competitions Conducted on Account of Silver Jubilee Celebration of Yenepoya Medical College
23 Jul 2024


Yenepoya Medical College Conducts Health Awareness Activities & Competitions in the Neighboring Schools as Part of the Silver Jubilee Celebration
19 Jul 2024

Yenepoya Medical College Conducts Health Awareness Activities & Competitions in the Neighboring Schools as Part of the Silver Jubilee Celebration
18 Jul 2024

Yenepoya Deemed-To-Be-University Signs MoU with Emversity School of Allied Health Sciences to Offer Work-Integrated Degree Programs
14 Jul 2024



Yenepoya Medical College Hospital Celebrated National Doctor's Day 2024 by Honouring the Exemplary Contributions of the Doctors
01 Jul 2024

21st Graduation Day Ceremony of Yenepoya Medical College on 19 May 2025 from 03:00 PM onwards
19 May 2025

Centre for Ethics is hosting a 2 days workshop on "Climate Change and Gender" on 2nd & 3rd June 2025.
13 May 2025

International Youth Conference ICON YOUTH 2025 on May 15 th and 16 th 2025 at Yendurance Zone, Yenepoya (Deemed to be) University, Mangalore, in collaboration with the Ministry of Youth affairs and Sports, Government of India.
06 May 2025
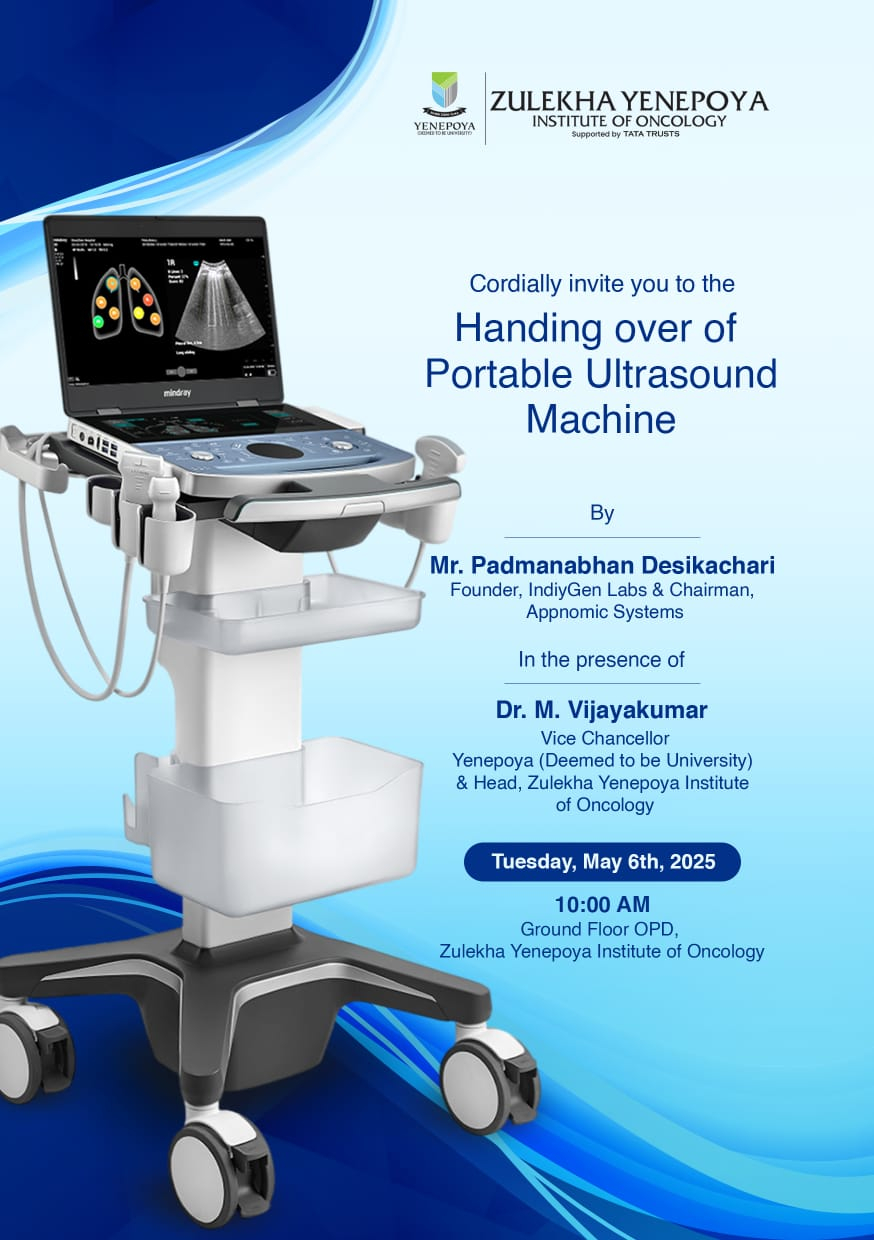
Zulekha Yenepoya Institute of Oncology Invites to the 'Handing over of the Portable Ultrasound Machine'
05 May 2025


Professors Dr. Anurag Bhargava and Dr. Madhavi Bhargava of Yenepoya Medical College have contributed to the groundbreaking paper, "A roadmap for integrating nutritional assessment, counselling, and support into the care of people with tuberculosis," published in Lancet Global Health
28 Mar 2025

Join Racefor7 - Run for Rare Diseases on 23 February 2025, at Yendurance, Yenepoya Campus
19 Feb 2025

Library e-Resources and Research Tools , How to Find Suitable Indexed Journals and Book Publishers for Publications Without APC
18 Feb 2025

The National Service Scheme (NSS) of Yenepoya (Deemed to be University) in association with the National Integrated Forum of Artists & Activists proudly presents KALASIRI - a vibrant celebration of art and culture
12 Feb 2025


Free Cancer Awareness Program & Screening Camp by Yenepoya (Deemed to be University) at Mangalore Beach Festival
30 Jan 2025

Yenepoya (Deemed to be University) is one of the Proud Partners of Mangaluru Beach Festival from 31st January to 2nd February
30 Jan 2025

Invitation to attend Session 4 - Career Counselling Series : Climate change and health
14 Jan 2025

Sensitization and User Awareness Program on 'Knimbus: Off-campus, E-Resources Access Tool'
19 Dec 2024

Sensitization and User Awareness Program on "'Edzter: Online Newspapers and Magazines database"
18 Dec 2024

An Al-powered Tool for Enhancing the Quality of Academic and Professional English Writing
12 Dec 2024

Sensitization and User Awareness Program on "ClinicalKey Flex (Physician): Full-text Health Science Database
11 Dec 2024




Yenepoya School of Engineering & Technology Observes 'World Science Day for Peace & Development'
10 Nov 2024






Yenepoya Dental College has been awarded the 'Best in Asia' by Pierre Fauchard Academy, US - Recognition by A Global Authority in Dentistry.
30 Apr 2025

Yenepoya Dental College Shines at the Pierre Fauchard Academy International Conference and Awards 2025
28 Apr 2025

Yenepoya (Deemed to be University)'s ACTS Yen, Simulation Center Accredited by ASPiH
12 Feb 2025

The Sustainability & Green Initiatives of Yenepoya Medical College Hospital and Yenepoya (Deemed to be University) is Published in the NABH Guidebook for Climate Action & Sustainability in Health Care
04 Feb 2025

Yenepoya (Deemed to be University) and Teachspoon EdTech Pvt. Ltd. Forge Strategic Partnership to Revolutionize Medical and Dental Education
12 Dec 2024


Pediatric Kidney Transplant Performed successfully at Yenepoya Medical College Hospital
30 Oct 2024

{kind=link}

MOU Between Yenepoya (Deemed to be University) and Bankers Institute of Rural Development (BIRD), Mangaluru
18 Sep 2024
ACADEMICS

-

Yenepoya Medical College
-

Yenepoya Dental College
-

Yenepoya Nursing College
-

Yenepoya School of Allied Health Science
-

Yenepoya Pharmacy College & Research Centre
-

Yenepoya Institute of Arts, Science, Commerce & Management
-

Yenepoya Ayurveda Medical College & Hospital
-

Yenepoya Homeopathic Medical College & Hospital
-

Yenepoya Naturopathy & Yogic Science college & Hospital
-

Yenepoya Physiotherapy College
-
Yenepoya Medical College & Hospital
-

Yenepoya School of Design
-

Yenepoya School of Engineering & Technology

Rankings & Accreditations

A+ GRADE
National Assessment and Accreditation Council 2020.

Ranked 95
University has been ranked 95 among the top 100 universities in the country.

NABH
NABH has been assessed and found to comply with NABH Accreditation Standards for Hospitals.

NABL
NABH has been assessed and found to comply with NABH Accreditation Standards for Hospitals.
WHAT'S YOUR INTEREST?
- Recent searches:
- UG
- PG
- Value-Added Courses
- PhD
- Fellowship
- Super Speciality
- Allied Health Science
- Certificate Course
- Diploma

more opportunities
to lead
“Yenepoya (Deemed to be University) brings you a world-class educational experience in the pristine surroundings of a tranquil south Indian town. Nature is the closest companion on the campus that houses institutes offering the best in education. Come explore Yenepoya and evolve into a technology-enabled future.”
Apply now